Chapter 3 Methods
If there is only one batch in the input data, the online tool only take the data normalization step. If there are multiple batches in the input data, the online tool normalize the data in each batch first and then take the batch effect correction. Figure 3.1 shows the the general workflow of the tool.
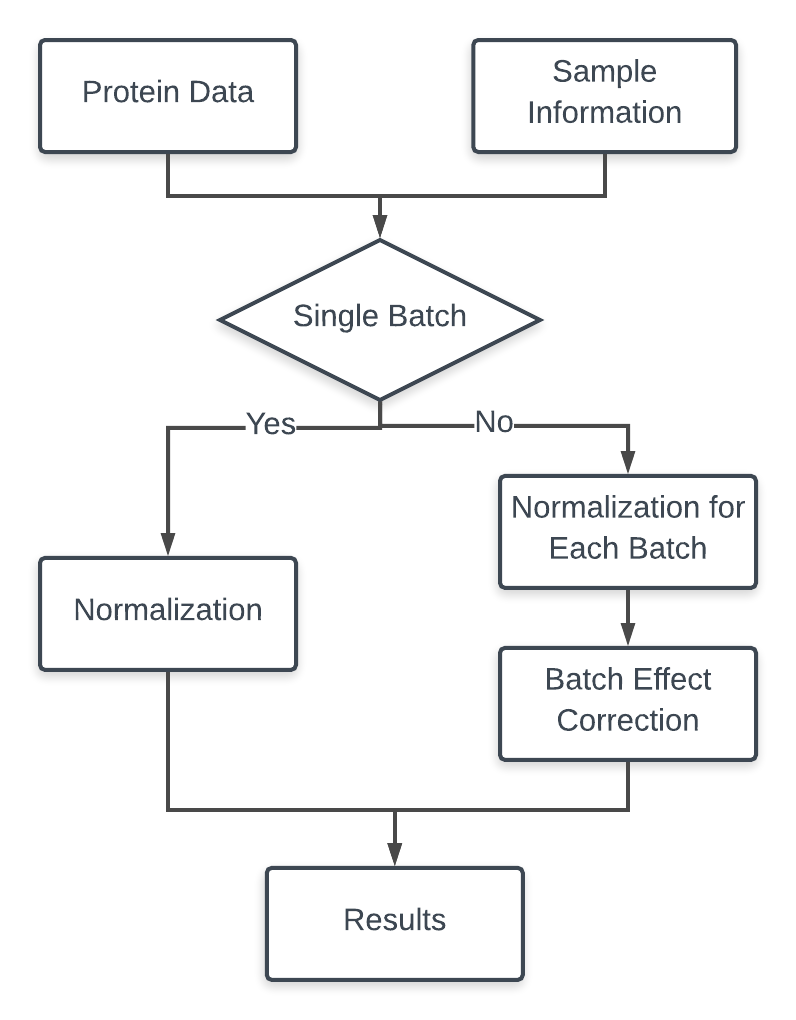
Figure 3.1: Workflow of the online tool
3.1 Single Batch
If the input data is in a single batch, the online tool only takes the normalization step. The following normalization methods are implemented in the online tool.
3.1.1 Sample Loading
This method assumes that the summation of abundance for all the proteins is the same for all the sample.
3.1.2 Median
The median normalization is based on the assumption that the median of protein abundance is the sample for all the samples.
3.1.3 Quantile
In quantile normalization, the quantile of each sample is replaced by the mean of the same quantile of all the sample (Bolstad et al. 2003).
3.2 Multiple Batches
If the input data contains samples from multiple batches, the online tool will take normalization step for each batch first and then remove the batch effect between different batches. Now we have implemented the following batch effect correction methods in the tool.
3.2.1 ComBat
We used ComBat method in R package sva. The details of the method can be file in the paper (Johnson, Li, and Rabinovic 2007).
3.2.2 Limma
Function removeBatchEffect in R package limma (Ritchie et al. 2015) is used to remove the batch effect.
3.2.3 IRS
This method is based on the same pooling sample across different batches. It assumes that the pooling samples have the same protein abundance at different batches and calculates a scaling factor from this assumption. The details of this method can be found at (Plubell et al. 2017).